Alfredo Prof. Brusco
Meccanismi patogenetici nella leucodistrofia autosomica dominante e possibili approcci terapeutici
Le leucodistrofie sono una famiglia eterogenea di rare patologie genetiche caratterizzate da una degenerazione progressiva della sostanza bianca cerebrale. Le forme più frequenti di leucodistrofia si manifestano in età infantile o nella pubertà con eredità autosomica recessiva (es. Krabbe disease) o recessiva legata all’X (es. adrenoleucodistrofia). La leucodistrofia autosomica dominante dell’adulto (ADLD) è una forma che si manifesta nella 4a-5a decade di vita. Questa patologia progredisce in maniera lenta ed è letale; clinicamente è caratterizzata da un esordio con disturbi del sistema nervoso autonomo, disfunzioni piramidali e cerebellari ed alterazioni simmetriche della mielina a livello del sistema nervoso centrale. Dal punto di vista genetico, ADLD è causata dalla duplicazione del gene LMNB1 (Padiath et al., 2006), che codifica per un componente della lamina nucleare. Lamina B1, tre le sue funzioni fisiologiche, regola la struttura della cromatina, la trascrizione, la replicazione e la riparazione del DNA (Dechat et al., 2010).
L’ADLD è associata a livelli aumentati di messaggero e proteina Lamina B1. In vitro, l’aumentata espressione di LMNB1 causa un’alterazione nel differenziamento degli oligodendrociti (Lin and Fu, 2009). Grazie a una collaborazione internazionale abbiamo raccolto e studiato dal punto di vista molecolare la più ampia casistica di pazienti ADLD ad oggi descritta, un totale di 20 famiglie provenienti da tutto il mondo. Questo studio ci ha permesso di confermare che la sola duplicazione del gene LMNB1 causa l’ADLD, che le duplicazioni sono nella maggior parte causate da micromologia, e che non sono ricorrenti. In tutti i pazienti analizzati, sia il messaggero sia la proteina Lamina B1 sono risultati aumentati rispetto a controlli sani, confermando come l’ADLD sia causata da un accumulo di questa proteina, che nel cervello dei pazienti è associato alla degenerazione progressiva della sostanza bianca.
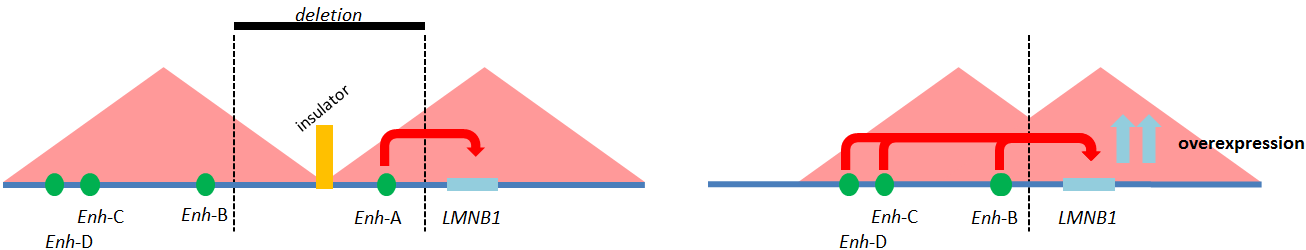
Il nostro gruppo ha anche descritto una famiglia italiana di cinque generazioni (ADLD-1-TO) affetta da una forma “variante” di ADLD, con caratteristiche cliniche e neurologiche sovrapponibili a quelle riscontrate nelle famiglie con duplicazione del gene LMNB1 (Brussino et al., 2010). Anche in questa famiglia la malattia è causata dall’aumentata espressione del gene LMNB1, in assenza, però, della duplicazione del gene stesso. Abbiamo recentemente dimostrato che in questa famiglia l’ADLD è causata da un meccanismo noto come “enhancer-adoption”: una delezione a monte del gene LMNB1 elimina un insulator ovvero una barriera di cromatina che normalmente impedisce l’interazione non fisiologica tra elementi regolatori e geni localizzati nelle vicinanze (Fig. 1). Questa eliminazione rende possibile l’interazione positiva tra tre elementi enhancer e il gene LMNB1, con conseguente aumento dell’espressione del gene stesso. Questa famiglia è il primo caso di “enhancer-adoption” in una malattia neurodegenerativa (Giorgio et al., 2015) e rappresenta un esempio di come malattie mendeliane, normalmente associate a una determinata mutazione in uno specifico gene, possano anche derivare da effetti posizione, ovvero da mutazioni che alterano i livelli di espressione di un gene, mantenendo però intatta la sua sequenza codificante. Queste mutazioni sono estremamente difficili da identificare nella pratica clinica perché coinvolgono sequenze di DNA normalmente non studiate.
Figura 1 Meccanismo di “enhancer adoption” in una forma di ADLD.
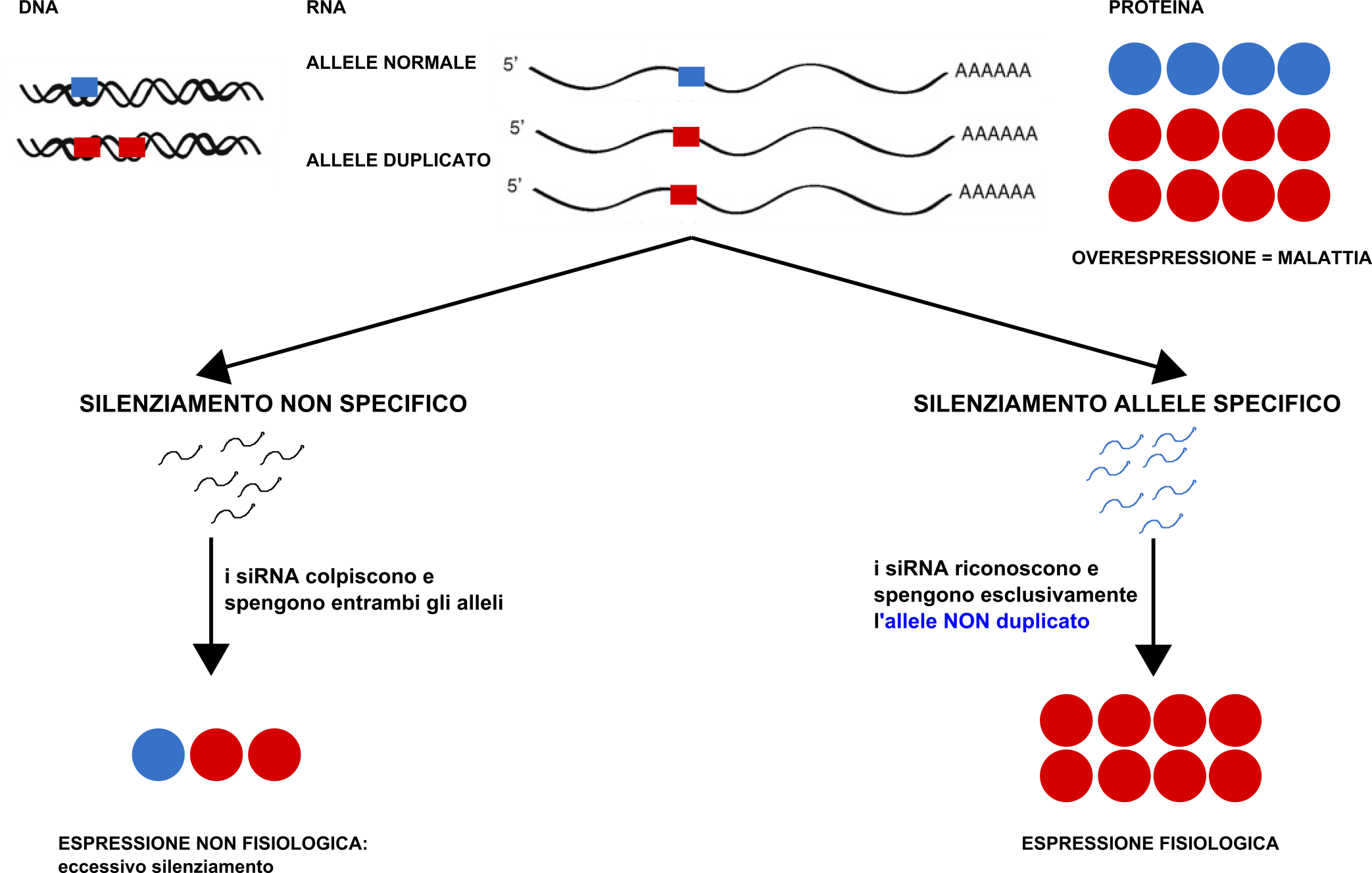
Ad oggi non è stata identificata alcuna terapia per l’ADLD. Il nostro gruppo ha deciso di intraprendere la strada del silenziamento allele specifico, una tecnica in grado di “spegnere” in maniera selettiva una singola copia di un gene. Questa strategia è stata utilizzata con successo per trattare malattie genetiche dominanti negative come l’Huntington e l’Alzheimer.
Ci aspettamo che il silenziamento allele specifico della copia in eccesso del gene LMNB1 riporti a livelli fisiologici l’espressione del gene, evitando l’accumulo della proteina Lamina B1 nel cervello dei pazienti e la conseguente degenerazione della sostanza bianca.
Figura 2. Schema riassuntivo dell’approccio “silenziamento allele specifico” per la terapia dell’ADLD.
Gruppo di ricerca
