Alfredo Prof. Brusco
Studio dei meccanismi patogenetici dell’atassia spinocerebellare 28
L’Atassia SpinoCerebellare 28 (SCA28) è una patologia autosomica dominante caratterizzata da atassia cerebellare progressiva e difetti oculomotori. Il gene mutato AFG3L2 (18p11.21) codifica per la proteina omonima, che compone, assieme a SPG7 (proteina mutata nella paraparesi spastica di tipo 7) il complesso esamerico mitocondriale m-AAA proteasi (ATPases Associated with a variety of cellular Activities). Entrambi gli isoenzimi (omoesamero di AFG3L2 e eteroesamero AFG3L2-SPG7) sono localizzati nella membrana mitocondriale interna e svolgono funzioni di chaperone e controllo qualità delle proteine mitocondriali (Fig.1).
Figura 1. AFG3L2 gene e proteina.
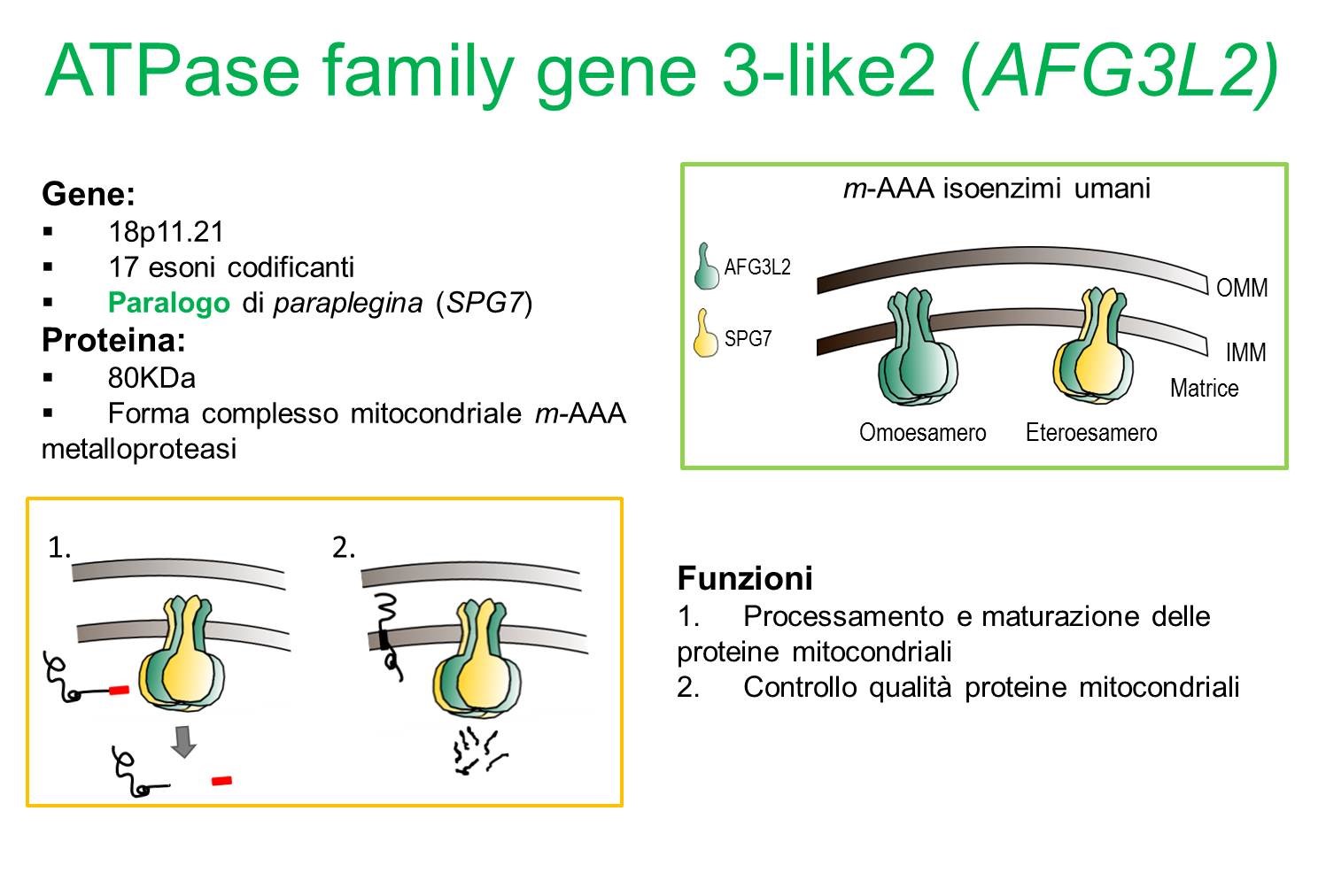
Le mutazioni in AFG3L2 identificate finora nei pazienti SCA28, sono per lo più missense (ad oggi sono state descritte solo una mutazioni frameshift e due delezioni del gene, rispettivamente una parziale -esoni 14-16- e una totale) e colpiscono il dominio peptidasico M41 della proteina (Cagnoli et al. 2010; Di Bella et al. 2010). Recentemente, nuove mutazioni sono state identificate in diversi domini della proteina in pazienti con fenotipi clinici differenti: una mutazione è stata identificata nel dominio transmembrana associata ad un fenotipo di mioclono (Mancini et al. 2015); mentre una mutazione nel dominio ATPasico della proteina è stata recentemente identificata in un paziente con atrofia ottica dominante.
Ad oggi, la maggior parte delle conoscenze relative alla funzione fisiologica delle proteasi m-AAA e dell’effetto delle mutazioni SCA28 sulla loro attività, deriva da esperimenti su modelli di lievito o lo studio del modello eterozigote knockout (Afg3l2+/Emv66).
Nel nostro laboratorio abbiamo recentemente generato un modello murino knockin (KI), inserendo nell’ortologo murino la mutazione p.Met665Arg identificata nel paziente SCA28 con esordio clinico più precoce. Lo studio di questo modello ci permetterà di capire l’effetto di questa mutazione sul complesso m-AAA, analizzando più nello specifico i tessuti direttamente colpiti dalla malattia, altrimenti non accessibili.
I primi esperimenti condotti sul modello KI ci hanno permesso di verificare che la mutazione inserita causa un fenotipo atassico tardivo se in eterozigosi (KI-hz), mentre causa morte perinatale quando in stato di omozigosi (KI-ho). Il KI-hz mantiene una struttura cerebellare normale, senza atrofia né danno alle cellule del Purkinje.
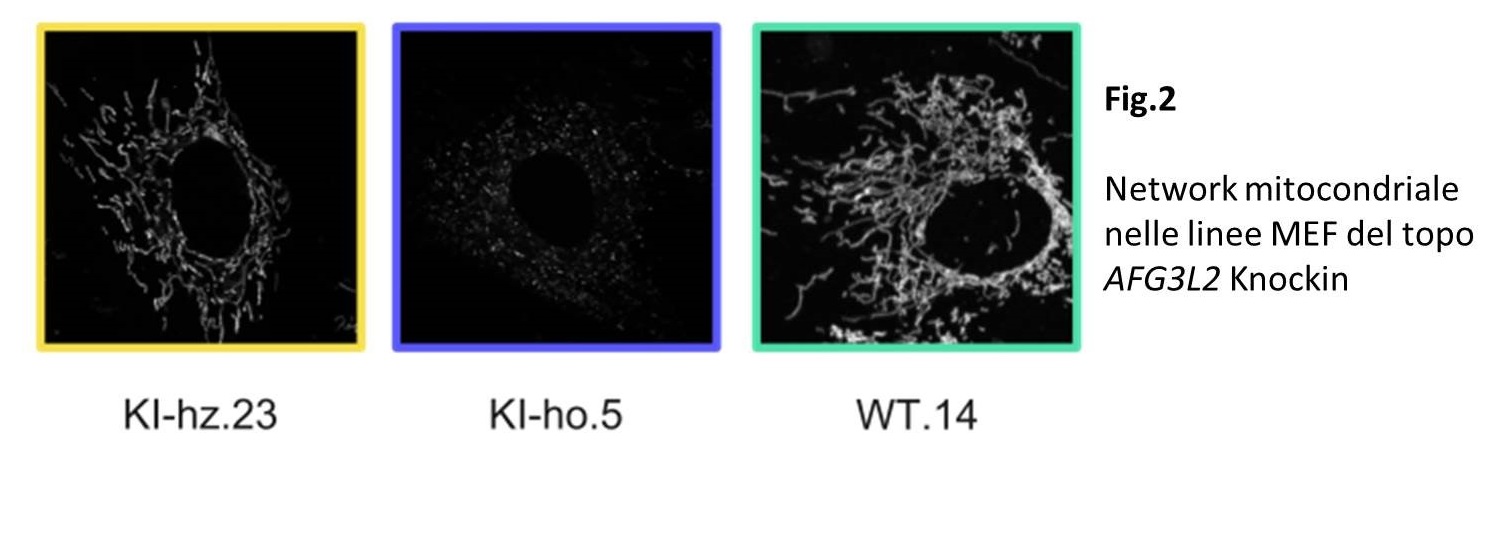
Analizzando più nel dettaglio i mitocondri, sede dell’attività del complesso m-AAA, abbiamo dimostrato che la mutazione p.Met665Arg in omozigosi causa una completa frammentazione del network mitocondriale mentre nei KI-hz il network mostra mitocondri intermedi-tubulari (Fig.2). Sembra inoltre evidente nei KI-ho una diminuzione del potenziale di membrana e l’analisi bioenergetica mostra una riduzione dell’attività del complesso III della catena respiratoria di circa il 25%, rispetto ai fibroblasti embrionali murini (MEF) di controllo (Fig.3).
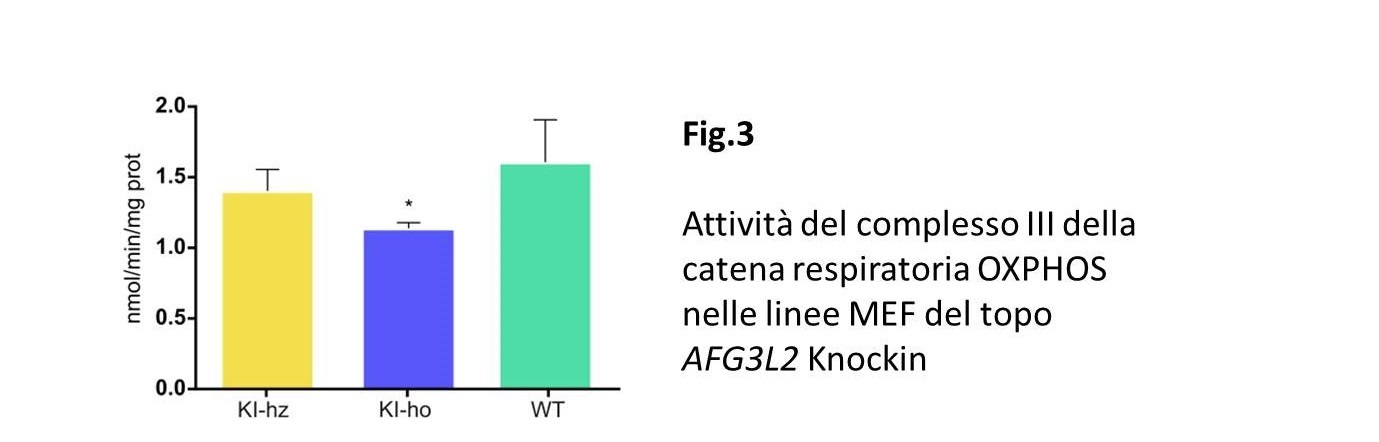
Questi risultati dimostrano che mutazioni a carico di AFG3L2 sono associate ad un lieve danno mitocondriale, e che probabilmente agiscono come ipomorfe, riducendo solo in parte la funzione della proteasi m-AAA.
Al momento stiamo valutando come altri meccanismi (autofagia/mitofagia) possano intervenire nella risoluzione dei danni mitocondriali in presenza del network mitocondriale frammentato visto nei KI-ho. Le conoscenze derivate da questi serviranno come punto di partenza per l’identificazione di potenziali nuovi target terapeutici.
Referenze
^Cagnoli C, Stevanin G et al: Missense mutations in the AFG3L2 proteolytic domain account for approximately 1.5% of European autosomal dominant cerebellar ataxias. Hum Mutat 2010, 31(10):1117-1124.
^Di Bella D, Lazzaro F et al: Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat Genet 2010, 42(4):313-321.
^Mancini C, Orsi L et al: An atypical form of AOA2 with myoclonus associated with mutations in SETX and AFG3L2. BMC Med Genet 2015, 16:16.
Gruppo di ricerca
